Multiscaling
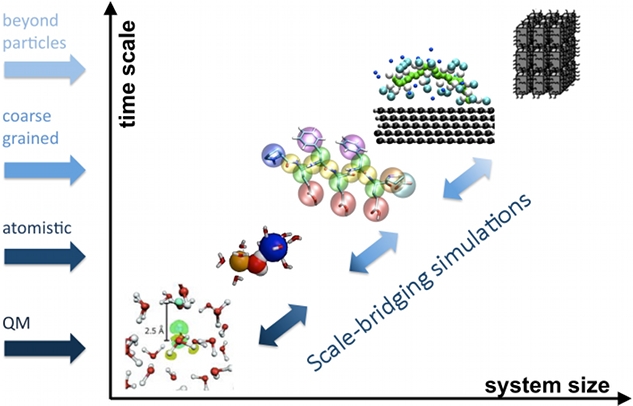
We develop systematic coarse graining methods, i.e. methods to derive models at a lower level of resolution based on information from simulations at higher, e.g. atomistic resolution.
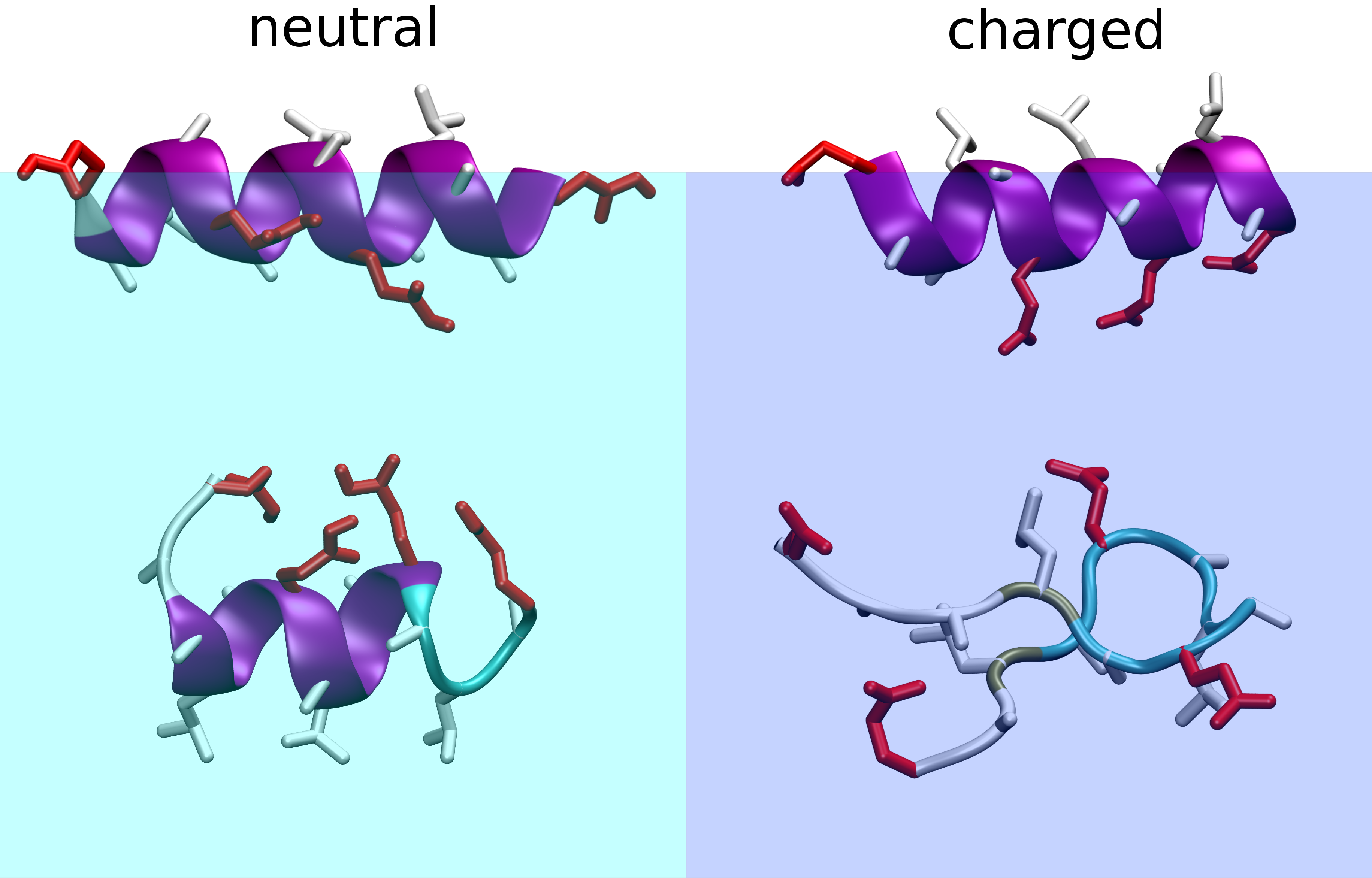
We carry out atomistic and coarse grained studies of environmentally driven folding. Amphiphilic peptides respond to the presence of interfaces, pH-changes or other changes in the solution composition by conformational transitions. Such peptides are for example initially designed as cell penetrating agents for drug targeting. Atomistic simulation studies can accompany experiments from collaborators to understand the driving forces for different folds and folding transitions. Moreover we investigate how coarse grained models can be designed such that they reproduce such responses to environment changes.
In recent years, machine learning techniques have become very popular and surround us in our daily life already. They help us to rank internet search results, enable software to read hand writing, recognize voice commands, and sort out spam emails. Because of the huge data amounts produced in Simulations, computational chemistry is ideal for the application of machine learning. In this project machine learning techniques are used to develop coarse grained molecule representations.
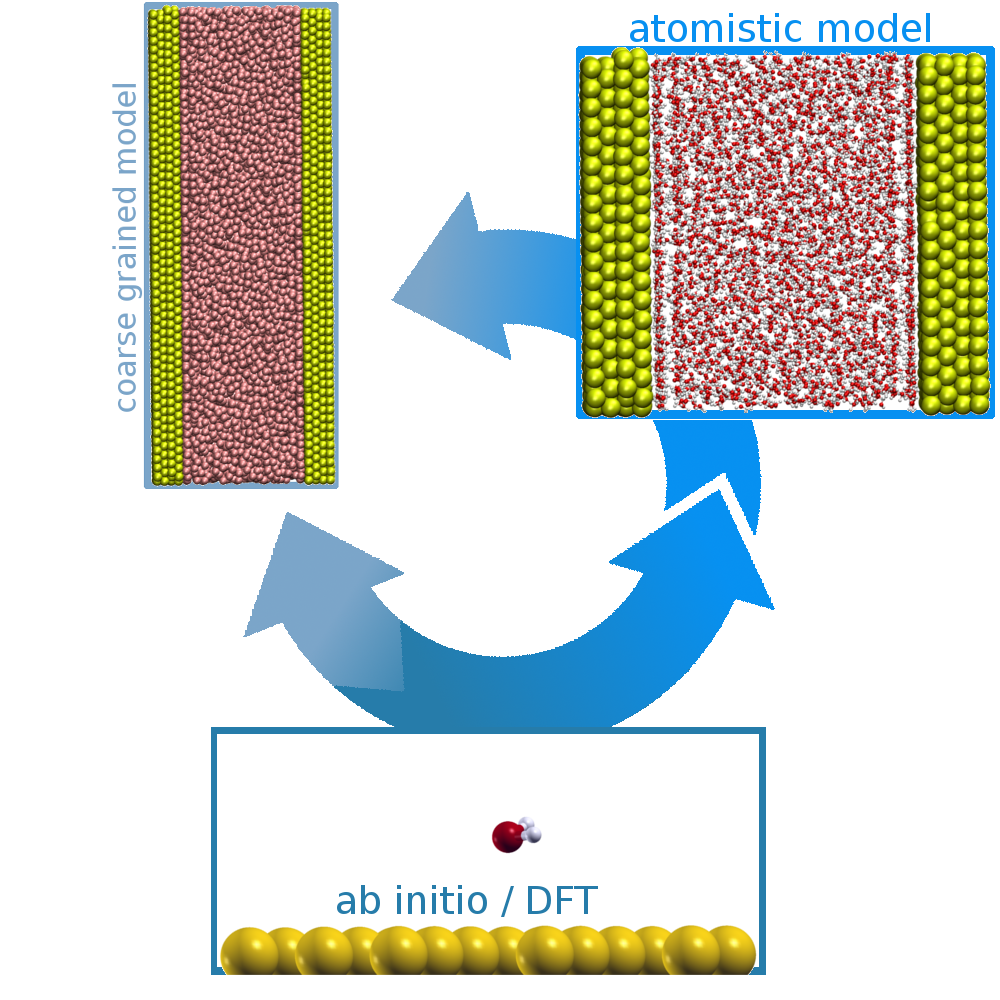
Systems of heterogeneous mixtures are important for various research fields. We are interested in the behaviour of large systems like bio-macromolecules in contact with gold surfaces or particles. Therefore, we use density functional theory calculations to obtain reference for optimisation of classical model parameters which will be used for MD simulations in the future.
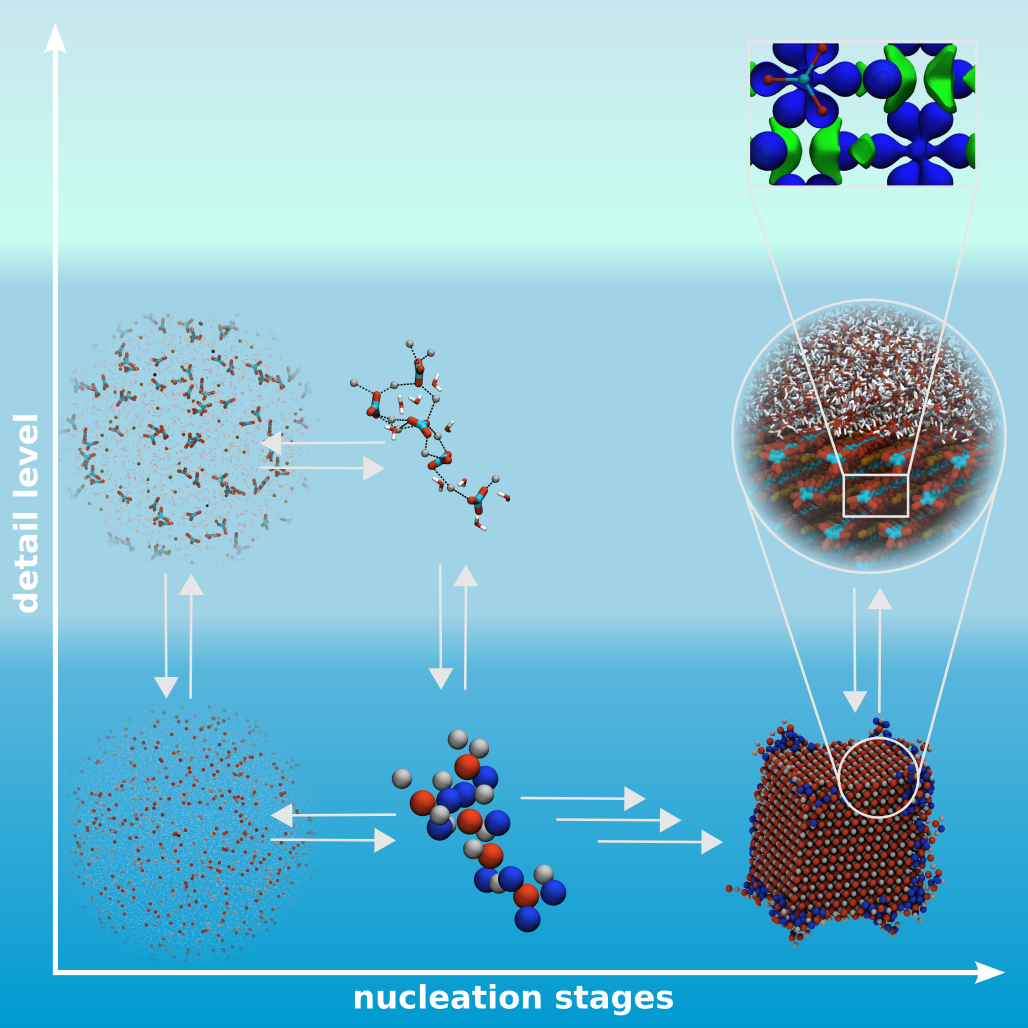
Nucleation as well as crystallization and growth of nuclei are important steps in numerous processes and applications, found in the inanimate and animated world. It could be shown that these processes can follow different pathways, classical and non-classical nucleation, which can be addressed by MD simulations. However, the different stages of these processes are on complete different time and size scales. Therefore, studies on different resolution levels, coarse-grained, atomistic and quantum mechanic levels, are needed and have to be linked to give a insight view in the underlying mechanisms.
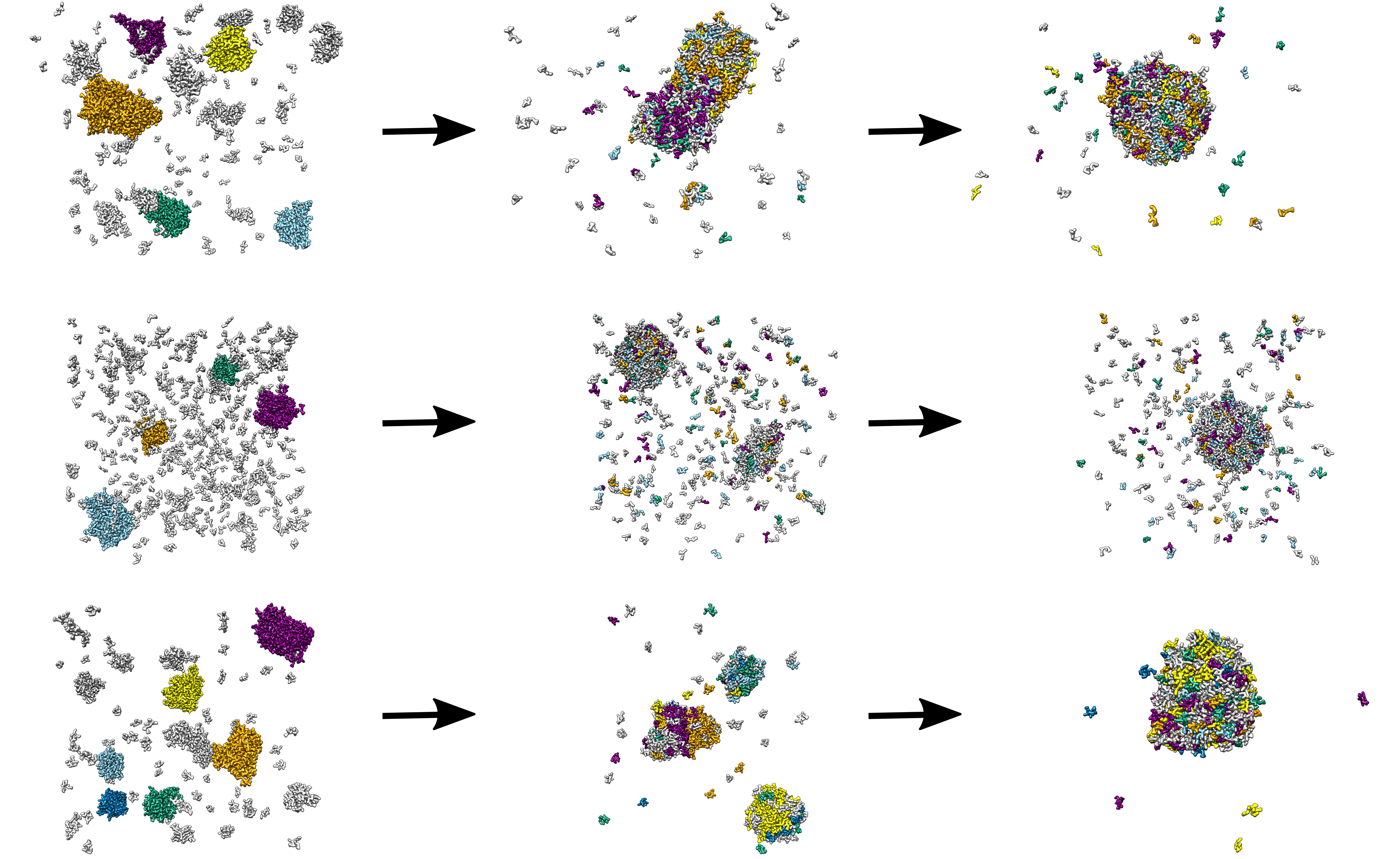
Development of new well ordered, functional biomaterials based on the underlying principal of self assembly has immense application in nanotechnology, nanomedicine and tissue engineering. Peptide based nano-materials are not only biocompatible but also their properties can be altered easily by slight changes in environmental conditions and/or side-chains of amino-acids. We are doing multiscale simulation study on many interesting peptides systems that exhibit different morphologies upon slightly altering the primary sequence of peptides.
J. Chem. Theory Comput. 2019, 15, 2, 1453–1462 doi
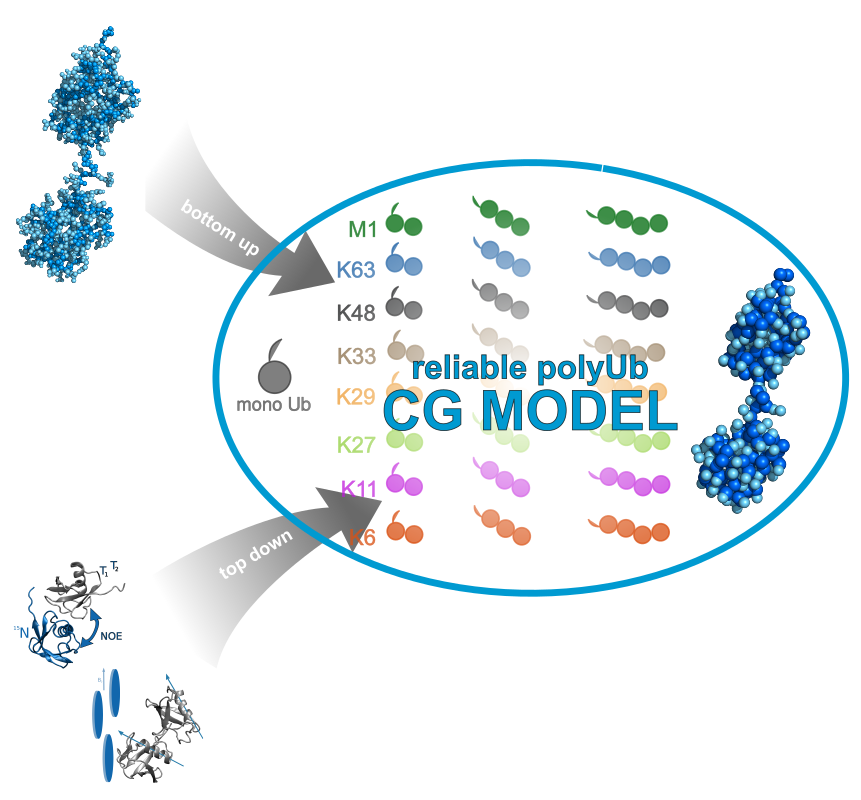
The ubiquitin system is involved in a variety of eukariotic cellular processes. It includes ubiquitilation as a post translational modification where substrate proteins are decorated with a single ubiquitin but also with chains containing several of these proteins. Different capabilities of linkage positions between distinct ubiquitin units provide variable chains. Hereinafter, ubiquitilated proteins are recognised by chain type specific antibodies and get processed by regulatory mechanisms like degradation but also by signalling and trafficking. Underlying recognition mechanisms will be elucidated by a combination of molecular modelling experiments facilitating interpretation of experimental investigations on the atomic scale.