Solid-softmatter interfaces

Studying the behaviour of a liquid phase in the immediate vicinity of solid interfaces is a particularly active research field for material science. Molecular modelling is an outstanding tool for this kind of systems since it allows microscopic insight on the atomistic scale which is required to understand the complex interplay between heterogeneous systems.
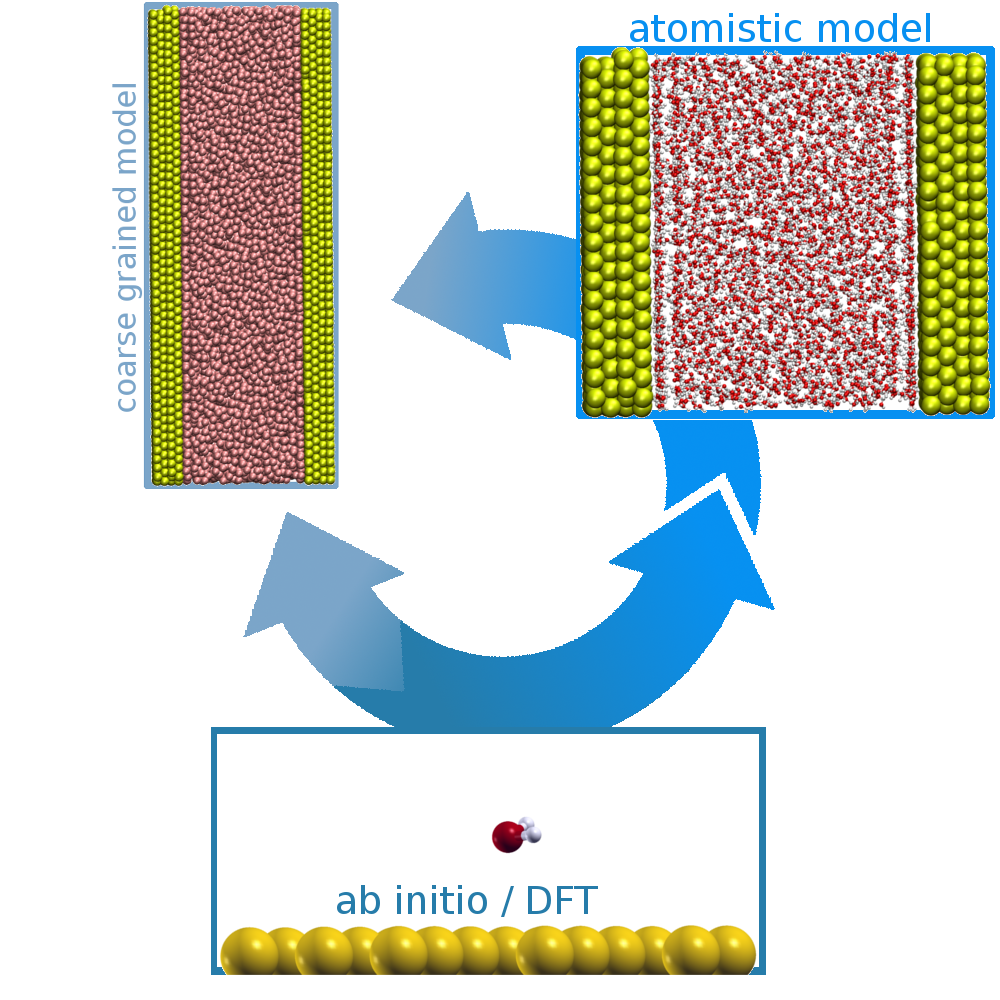
Systems of heterogeneous mixtures are important for various research fields. We are interested in the behaviour of large systems like bio-macromolecules in contact with gold surfaces or particles. Therefore, we use density functional theory calculations to obtain reference for optimisation of classical model parameters which will be used for MD simulations in the future.
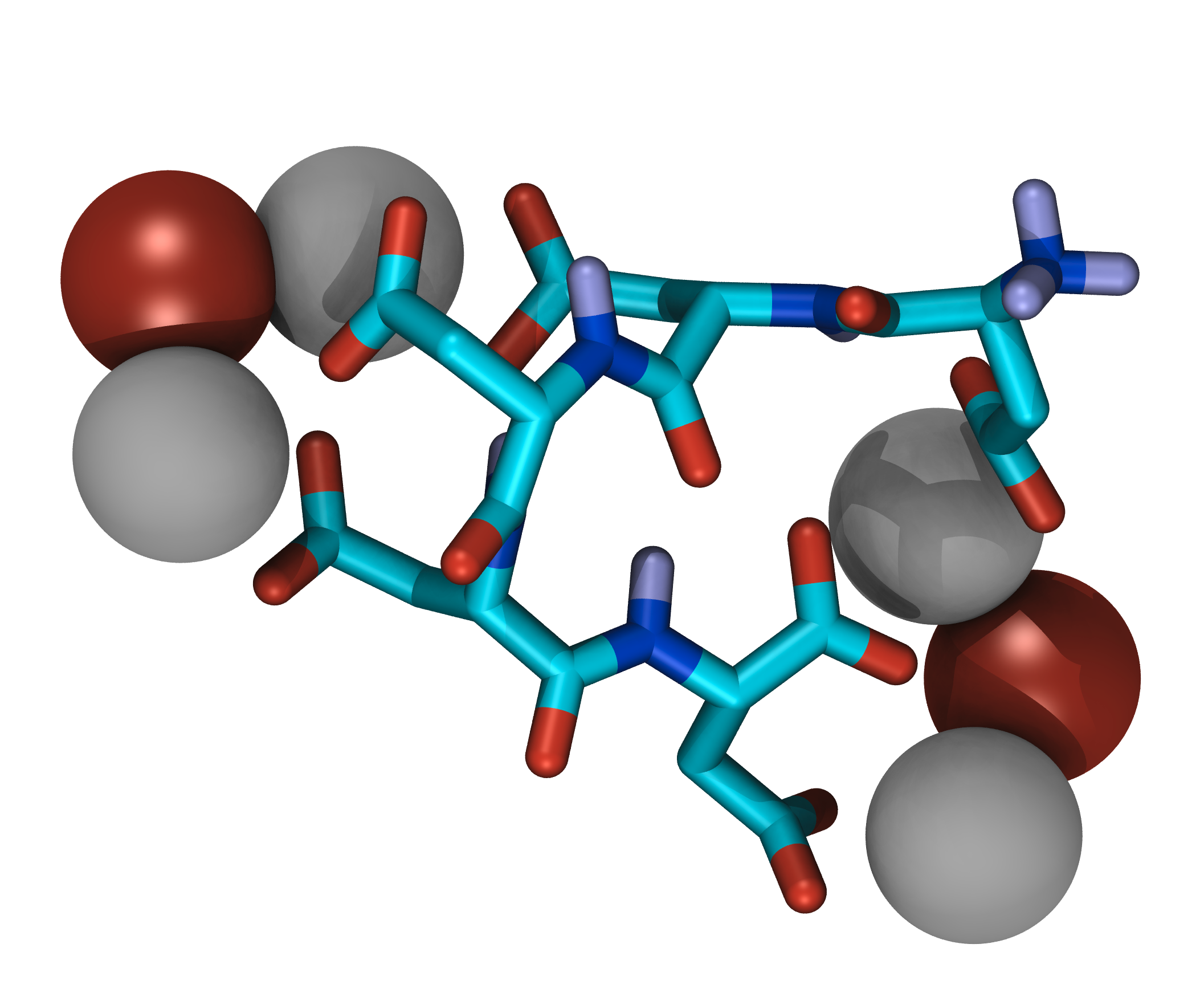
Some of the main components of biominerals besides the mineral itself are macromolecules, which appear to be the key for understanding their structural variety. Macromolecules isolated from calcium carbonate biominerals typically contain parts rich in aspartic acid. The influence of this amino acid on mineral formation can be explained with the ability to bind to metal ions with the negatively charged carboxylate group in the side chain.
Simulations of such mineralization modifiers suffer from slow sampling due to high energetic barriers caused by strong ion bridges. Even with the application of enhanced sampling techniques like Hamiltonian replica exchange and Metadynamics sampling to convergence remains a challenge. To tackle these sampling problems, the application of machine learning techniques is evaluated.