Conjugated Metal-Organic π-Systems
Alkenyl Ruthenium Complexes
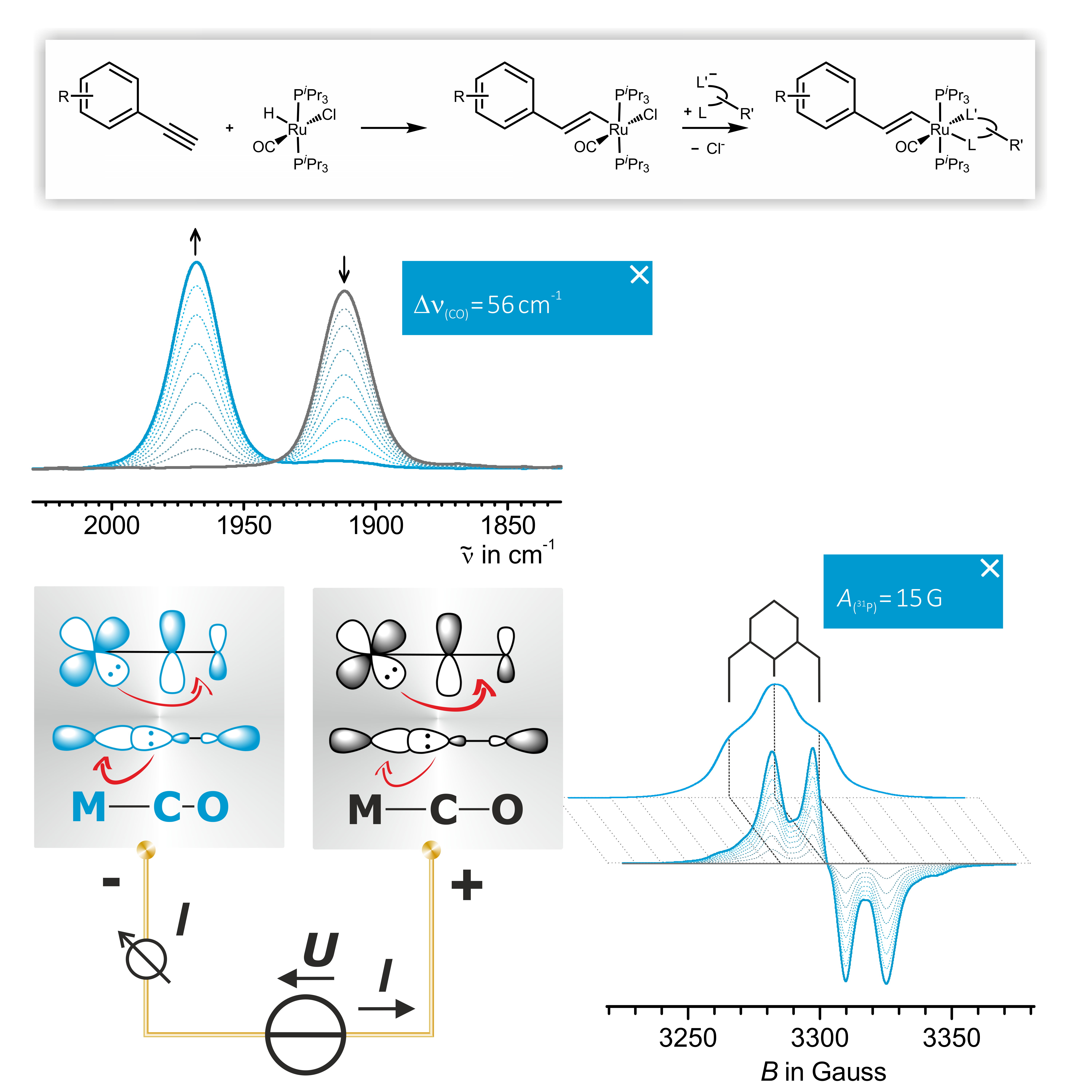
Alkenyl Complexes combine convenient atom economic syntheses with usually reversible oxidation of the alkenyl ruthenium electrophore. Monitoring the oxidation-induced IR shift of the Ru(CO) band and the 31P hyperfine splittings of the associated radical cations by EPR spectroscopy provides detailed information of the metal versus ligand character of the oxidation. Such experiments are conducted by directly coupling in situ electrolysis in transparent thin layer cells with IR, UV/Vis/near infrared (NIR) or EPR spectroscopy or by chemical oxidation with a suitable oxidant. Our present research centers in this area centers around three fields.
Mixed Valency in Symmetric and Unsymmetric Alkenylruthenium Complexes
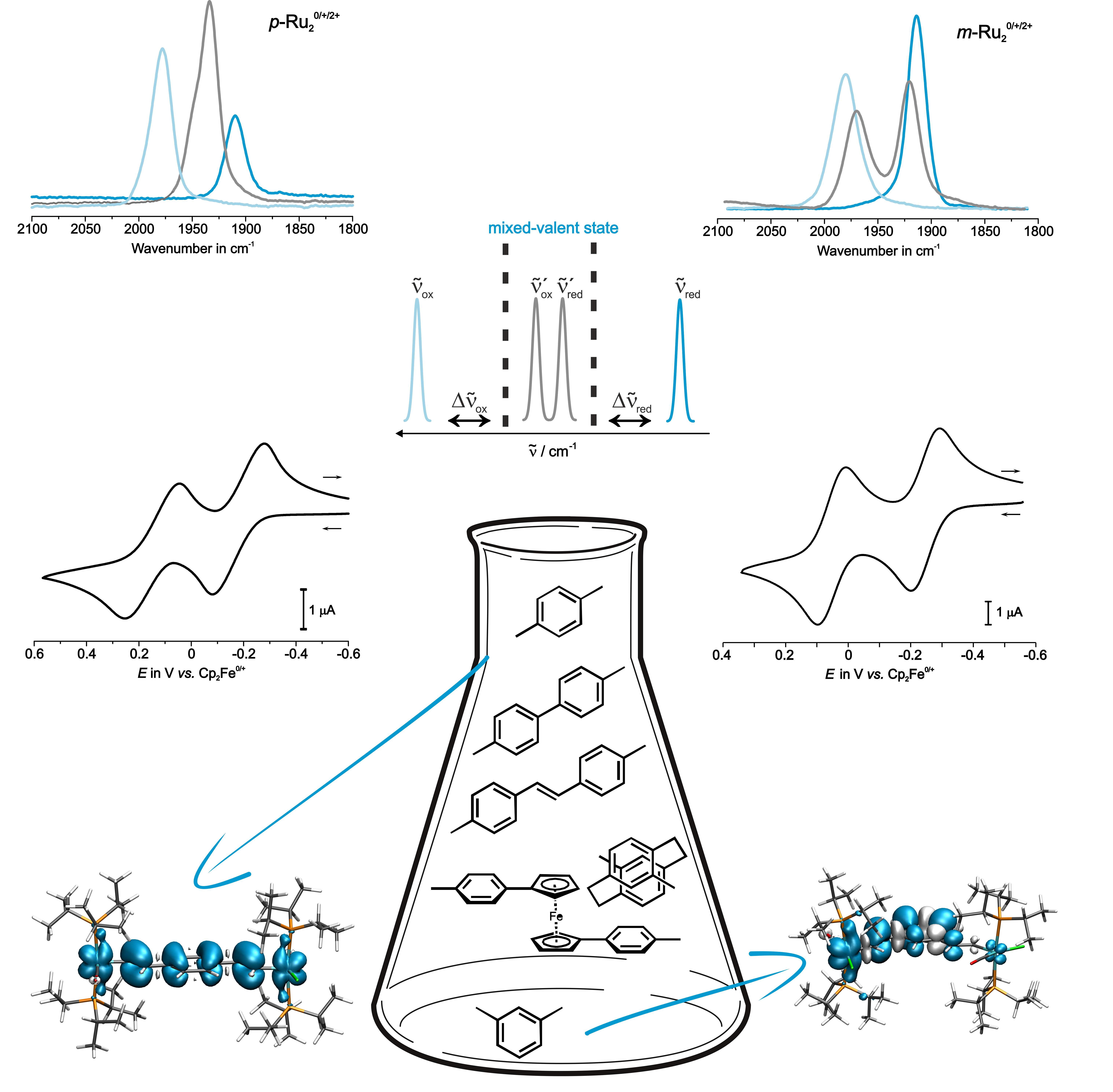
Complexes where two or more alkenylruthenium are connected to a common organic ligand typically feature one reversible oxidation for every CH=CH-{Ru} “substituent”. We aim at a detailed analysis of how the charge and the spin density distribute over the individual redox sites by experimental and quantum chemical means. We are particularly interested in the mixed-valent, odd electron states. For example, we have investigated the effects of lengthening the π-conjugated bridge between two { Ru(CO)Cl(PR3)2(CH=HC-)} entities from phenylene, to biphenylene (with various torsion angles) and stilbenyl. Some of the oxidized complexes combine the treats of strong electron delocalization and paramagnetism with two or more unpaired spins. This makes them likewise interesting for molecule-based electronics and as magnetic materials.
Derivatives with Au-binding functional groups are presently being tested by collaboration partners as molecule-based wires in single molecule conductance experiments. Further work in this area aims at complexes showing rapid electron transfer over long distances.
Radical cations of alkenylruthenium complexes featuring another, chemically different redox-active moiety may exhibit charge localization on just one redox site, or partial or complete charge and spin delocalisation over both dissimilar sites. We scrutinize conjugates of alkenylruthenium complexes with ferrocenes, triarylamines, carbazoles, triazatruxenes, porphyrins, squaraines or alkynyl metal complexes by combining electrochemistry and molecule spectroscopy in situ and ex situ approaches, and by quantum chemistry. In some cases, up to three isomers that differ in their charge and spin distributions (valence tautomers) coexist in equilibrium.
Alkenylruthenium Complexes as Electrochromic Materials
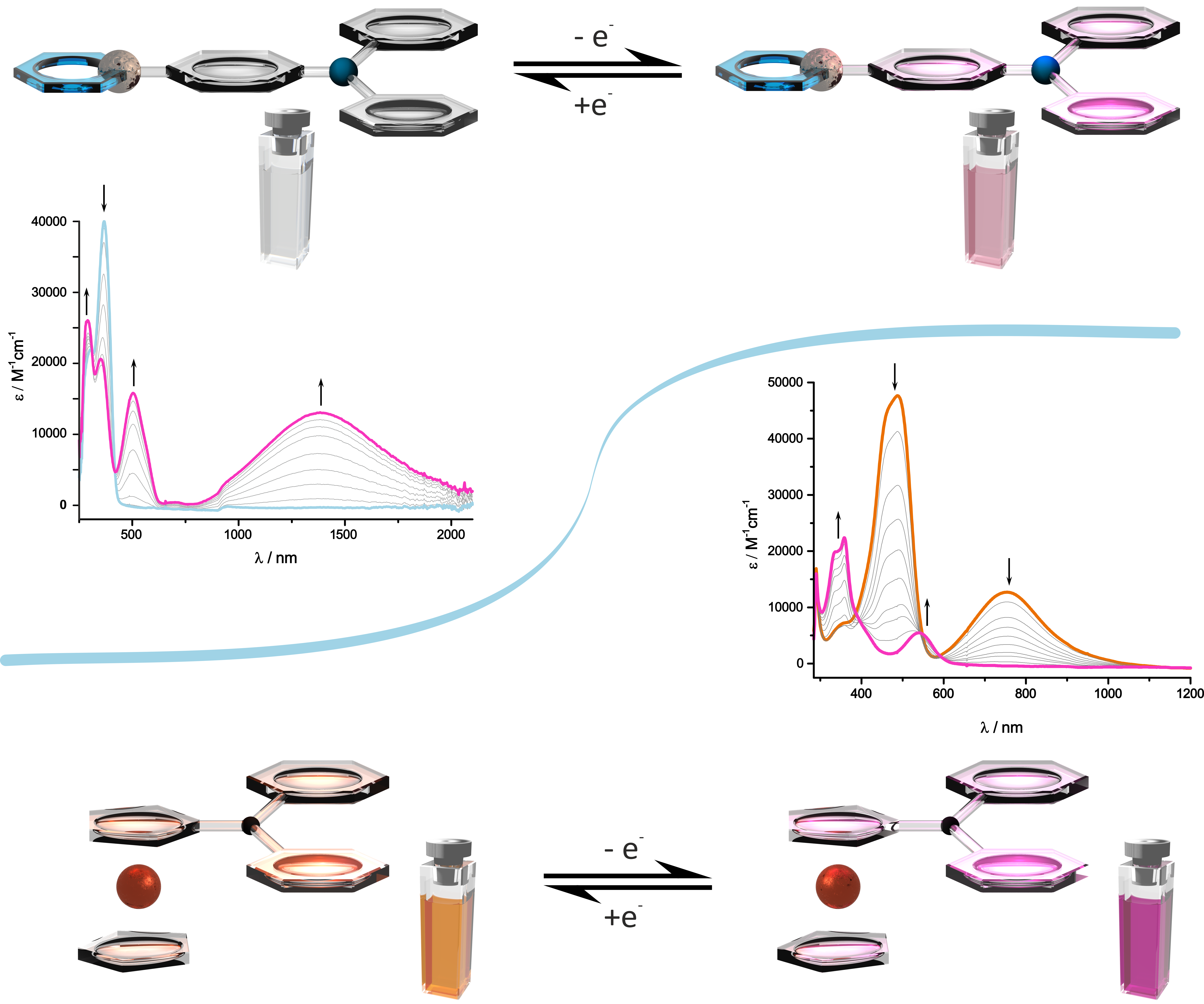
Oxidized alkenylruthenium complexes with two or more conjugated redox sites exhibit strong electronic absorption bands in the near infrared (NIR) or the spectral region at the Vis/NIR borderline which can be turned on or off by oxidation/reduction cycles. As their neutral parents are transparent in that spectral window these complexes constitute electroswitchable (poly)electrochromic dyes. Particularly high extinction coefficients at low energies are found for oxidized alkenylruthenium complexes with triarylamines, tetraphenylethenes, divinylarylenes, divinylstilbenes and squaraines. We presently explore pyrenes and extended heterocycles as constituents of such compounds. Electroswitchable NIR absorption is of great potential interest for heat transmission through glass windows or roofings, for controlling transmission through glass fibers, or for the construction of more efficient dye-sensitized solar cells.
Redox Active Ruthenium Metallamacrocycles
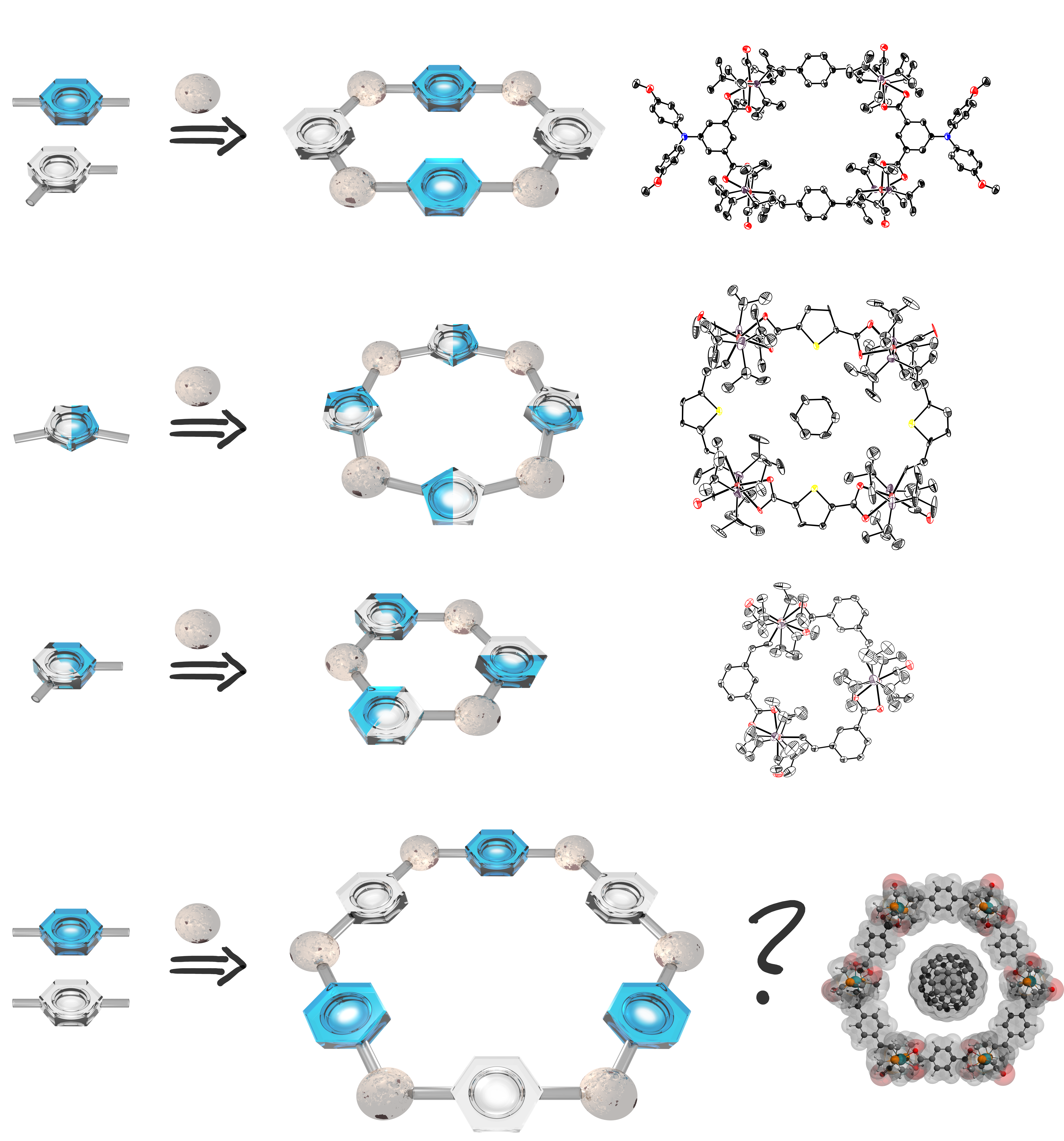
Styrylruthenium complexes with a chelating functionality at the aryl substituent or divinylphenylene-bridged diruthenium complexes and bis(carboxylates) with angular disposition of the carboxylate functionalities self-assemble to tri-, tetra- or hexaruthenium macrocycles. These structures are not only esthetically pleasing but exhibit rich electrochemistry and polyelectrochromism with strong absorption at low energy in the visible or in the near infrared of their oxidized forms. Moreover, the latter are paramagnetic with up to four independent spins. We presently aim at endowing these macrocycles with appropriate functionalities in order to use them as ligands or to access a further level of structural complexity through their self-association to higher-order structures. Further work is directed at exploring their host-guest chemistry towards suitable guests of complimentary shape or hydrogen-bonding properties or the fabrication of molecular conductive loops.
Pt Complexes with Covalently Attached Dye Ligands as Dual Fluorescence and Phosphorescence Emitters

Complexes of heavy-metal ions are known for their efficient intersystem crossing (ISC) in which generates long-lived and phosphorescent excited states. Owing to their long lifetime, the photoexcited states can also be used for triggering chemical reactions by electron or energy transfer to a suitable substrate. One major drawback of many of these complexes is that the underlying absorption band is often rather weak. Our approach to better sensitizers and emitters is to use strongly absorbing dyes as ligands and to directly attach them to platinum. We have prepared diimine platinum complexes where the dyes act as antennas for populating the emissive triplet state. On the other hand, direct π-bonding of a dye to the platinum ion also serves to populate a dye-based triplet state. As a result, such complexes exhibit dual fluorescence and phosphorescence emissions from the dye ligand. In particular, acceleration of the ISC from the T1 to the So state makes phosphorescence highly competitive to non-radiative deactivation processes, which results in high phosphorescence quantum yields. Examples are complexes with thioxanthones or BODOIPYs. Such dually emissive complexes are efficient one-component sensors for triplet quenchers, whose sensitivities can be tuned by the other coligands at the platinum ion. They also serve as efficient generators of 1O2 in photocatalyzed oxidation reactions. Further work is directed at widening the palette of usable dyes and at panchromatically absorbing platinum complexes with two different dye ligands with directional energy transfer from one dye ligand to the other.